
Research in the Surendranath Group focuses on the investigation and manipulation of chemical reactions occurring at solid-liquid interfaces. In particular, the Group aims to use electricity to rearrange chemical bonds by controlling interfacial reactivity at the molecular level. The chemistry of these interfaces is at the heart of nearly all contemporary challenges in renewable energy storage and utilization in a wide variety of devices ranging from batteries, to fuel cells, to electrolyzers and, therefore, addressing these challenges is essential for enabling a low-carbon energy future.
The macroscopic, device-scale, properties of any electrode are dictated principally by the individual elementary charge transfer reactions that occur at the solid-liquid interface. Thus, the development of next-generation devices for renewable energy storage and utilization requires strategies for understanding and manipulating these elementary charge transfer reactions at the level of atoms and molecules. The cycle of research in the group follows the following general trajectory: (1) novel interfacial structures are synthesized using tunable methods (2) the mechanisms of interfacial reactions are probed in detail to identify the efficiency limiting steps (3) the performance of each designer interface is evaluated and compared to identify new design principles for iterative optimization. This approach distinguishes itself from traditional methods for improving electrode performance that rely principally on trial and error.
Molecular Interfaces
The field of electrocatalysis has traditionally been divided into heterogeneous electrocatalysis, in which the active material is typically the metal surface of an electrode, and homogeneous electrocatalysis, in which the active material is typically a molecule in solution. Strong interactions between molecular active site and an electrode surface can blur this binary, giving rise to systems whose reactivity is distinct from that of the isolated molecule. We have extensively investigated Graphite-Conjugated Catalysts (GCCs) - formed from the chemical grafting of molecular active sites onto graphitic materials, we have shown that the strong electronic coupling between the extended solid and molecular active site engendered by the covalent linkage enables a fundamental change in catalytic reactivity, eliminating the need for the reaction to proceed through a redox intermediate (as is typical in homogeneous catalysis) and allowing instead for inner-sphere bond formation steps to occur in concert with electron transfer (as is typical in heterogeneous catalysis).
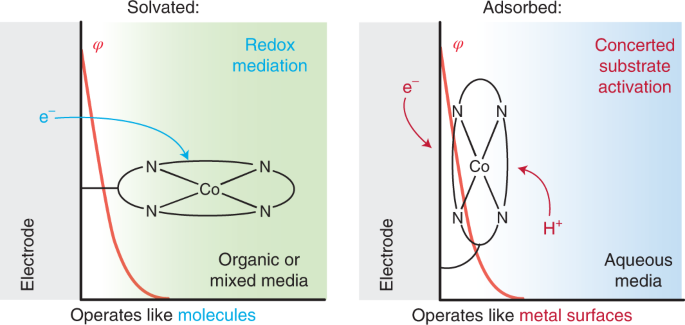
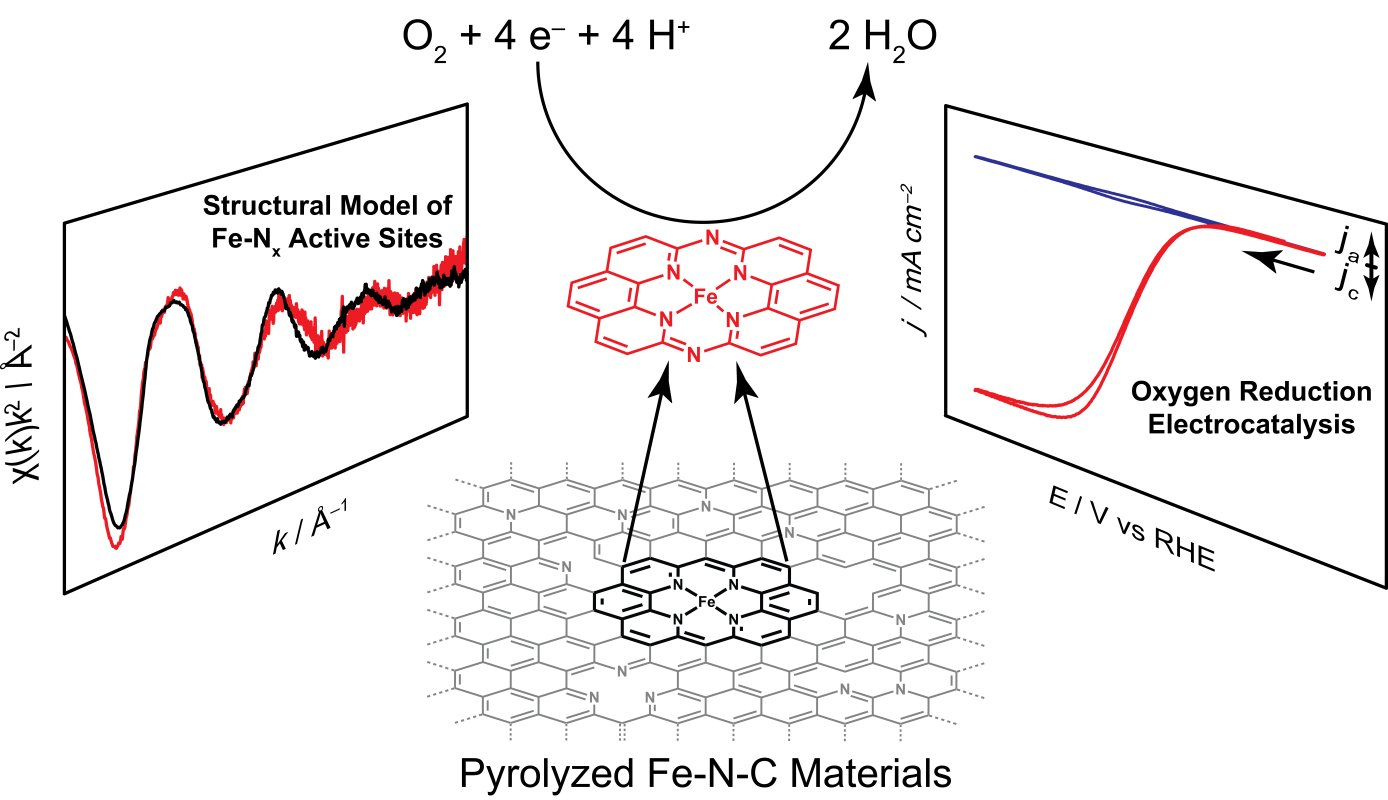
In addition, we have investigated the active sites of Iron- and Nitrogen- doped carbon (Fe-N-C) materials, a low-cost oxygen reduction catalyst, and discovered that these sites can be locally modeled by a molecular iron macrocycle with pyridinic coordination. Our group is currently in the process of extending our studies of molecular/electrode interaction to systems which lack direct electronic conjugation between the surface and active sites - for instance, we have recently shown that adsorbed metalloporphyrins exhibit metal-like reactivity during hydrogen evolution catalysis, in contrast to the free molecular form of the catalyst.
Electrochemistry in Exotic Media
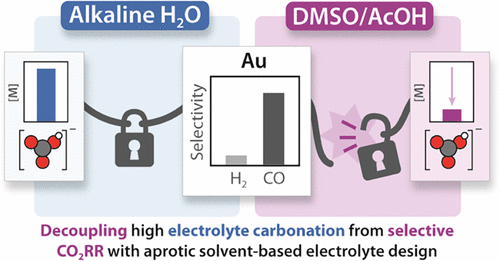
Working in diverse electrolytes and reaction conditions provides opportunities not available in typical electrochemical media. We consider optimization of electrolyte and the electrode/electrolyte interface as key parameters for research, that can enable new insights for any reaction of interest. In the case of carbon dioxide reduction, we found that combining DMSO with organic proton donors provides a non-aqueous reaction environment that enables favorable selectivities while circumventing electrolyte carbonation. In aqueous conditions, exchanging alkali carbonate electrolytes for weakly coordinating organic cations provided a platform for us to demonstrate that cation coordination is not required for CO2 activation. Our group is now interested in the capacity for alternative electrolytes to enable selective CO2 reduction.
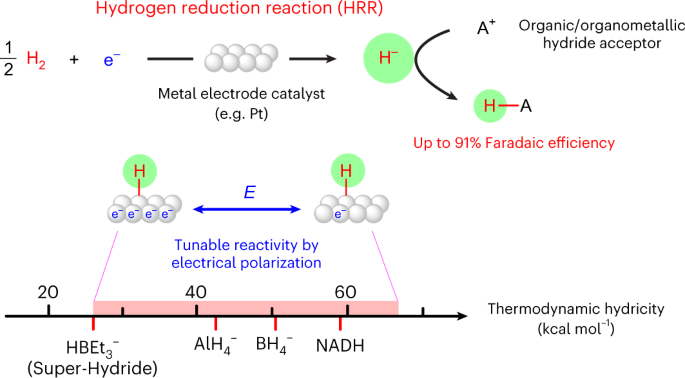
Alternative media also provide platforms for brand new reactivity. We recently developed a new electrochemical pathway with dihydrogen - the hydrogen reduction reaction. Using nonaqueous electrolytes with H2, we demonstrated that catalysts like Pt can catalyze the electrochemical reduction of H2 to generate reactive hydrides that can react with acceptor molecules in solution. We are currently investigating how this platform can be used to understand molecular hydricity, and to enable hydride transfer to interesting target molecules.
ElectroThermoCat
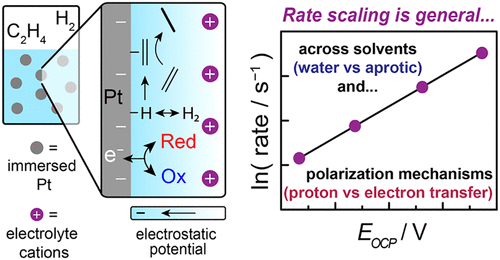
ElectroThermoCat is our catch-all term for the concepts that bridge the (traditionally separate) fields of electrocatalysis and heterogeneous "thermal" catalysis. These concepts come in a few distinct flavors, including but not limited to electrochemical promotion of thermochemical catalysis (so-called "EPOC" or "NEMCA" effects); thermal redox catalysis that proceeds via electrochemical mechanisms ("local short-circuit" mechanisms); and potential measurement in heterogeneous catalysis. In the realm of EPOC effects, we investigated ethylene hydrogenation under a variety of polarization conditions in both aqueous and nonaqueous conditions, unearthing a common dependence of rate on interfacial potential.
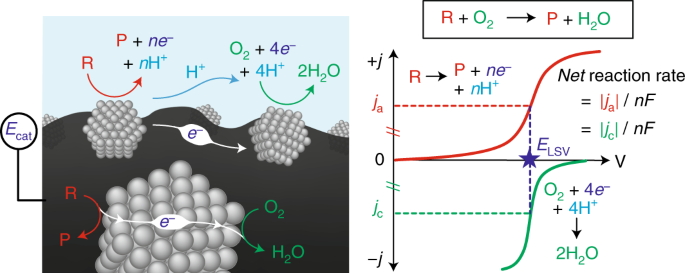
In redox catalysis, we have established a common framework centered around "corrosion-like" mechanisms that can precisely explain the rate and spontaneous catalyst polarization that occurs in quinone and alcohol oxidations. Our group is now interested in expanding our ability to measure potential in a variety of relevant catalytic contexts, and understanding how electrochemical polarization can drive many different classes of reactions.
Hypothesis-driven synthesis and rigorous physical characterization provide the basis for catalyst discovery and optimization. Students and postdoctoral scholars gain expertise in modern methods for the synthesis of inorganic coordination compounds, thin films, nanocrystals, and organic ligands. We employ a range of characterization tools including TEM, SEM, AFM, XPS, UV-Vis, IR, NMR etc. with a particular emphasis on electrochemical methods. These tools allow us to probe structure-function relationships that guide the development of new synthetic strategies.